The Protein Investigator
- Introduction
- Instructions
- Downloading and Installing
- A Sample Lab Manual
- Source Code
- Other Educational
Software by the author
(1) Introduction
Many lab exercises use molecular visualization software that allows students to to 'see' a protein
in its full 3-dimensional form and to make hypotheses about the interactions between various side
chains. However, these programs do not allow students to test their hypotheses because molecular
visualization software does not allow students to make changes to the protein sequence and observe
their effects on the protein's structure and function.
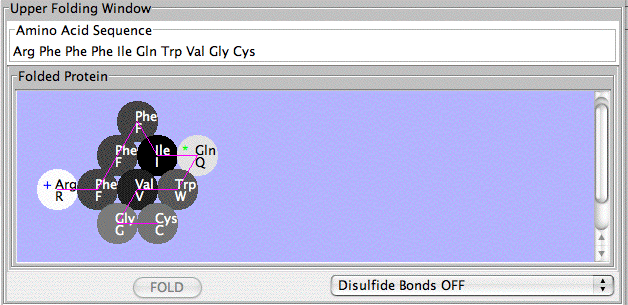
The Protein Investigator gives students an opportunity to interactively explore protein
structure and function in a simplified system. This is a highly-simplified model of protein folding.
It is not intended to predict the correct structures of any proteins; it is designed to illustrate
the major principles involved in that process. The important features of proteins that this software
retains are as follows:
- Amino acids have side-chains of varying hydrophobicity, charge, and hydrogen bonding
capacity.
- The amino acids a reconnected in an un-branched chain that can bend.
- Hydrophobic amino acids will tend to avoid the water that surrounds the protein;
hydrophilic amino acids will bind to the water.
- Amino acids that can form hydrogen bonds will tend to form hydrogen bonds if they can.
- Positively-charged amino acids will tend to form ionic bonds with negatively-charged
amino acids if they can.
- Like-charged amino acids will repel each other if they can.
- Ionic interactions are stronger than hydrogen bonds, which are stronger than
hydrophobic interactions.
- Proteins can be folded under oxidizing (Disulfide Bonds ON) or reducing
(Disulfide Bonds OFF) conditions.
Even though this software provides some important insights into protein folding, you should
always keep in mind that this is an approximation. The most important "gotcha's" to be aware of are:
- This program folds proteins in 2-dimensions only.
- This program treats all amino acids as equal-sized circles.
- This program folds the protein based on the interactions between the side chains only.
- This program does not model secondary or quaternary structure.
- This program assumes that all side chains with hydrogen bonding capability can bond with each other.
These simplifications are necessary for two reasons. The first is technical: it turns out to be
extremely difficult to predict the full 3-d folded structure of a protein given only its amino
acid sequence. As of the writing of this web page, it takes a super-computer several days to
predict the fully-folded shape of even a small protein like lysozyme. Even then, the predictions
don't always match known structures. The second reason is educational. As you saw in other labs,
proteins are complex 3-dimensional molecules. It can be hard to find your way around when inside one.
Likewise, it would be very difficult to visually compare two protein molecules to observe the effects
of changes to their amino acid sequence. It would be easy to miss the forest (the forces that control
protein structure) for the trees (the tiny details of the structures).
For these reasons, we will use this simplification. It retains the properties of amino acids that
are important in Bio 111 while being simple and fast.
You can view the help file included with Protein Investigator.
This software was written by Brian White (Biology Department;
UMass Boston), Ethan Bolker (Math/CS Department; UMass Boston), and
the MGX team.
(3) Downloading and Installing
In order to run the Protein Investigator, you will need the latest version of Java.
- Macintosh OS 9.2 and lower does not have Java; you cannot run Protein Investigator with this OS.
- Macintosh OS X has the proper version of Java built in. You can run Software Update to be sure
you have the latest version.
- Windows does not come with Java installed. You can download it free from
this link
You can get the latest version of the Protein Investigator
(Version 3.0.2 as of October 30, 2009)
from the appropriate link below:
(4) A Sample Lab Manual
- Lab manual in pdf format.
(5) Source Code
Protein Investigator is Open source and subject to the
GNU Public License.
The Source Code is available in the format
of an Eclipse project file.
This page maintained by Brian White.